bam文件如何打开查看 测序数据的格式解读方法
bam文件如何打开查看,Bam文件是高通量测序中的标准文件格式之一,存储了测序数据的信息。但是对于初学者来说,如何打开和查看Bam文件,以及如何解读其中的测序数据是一件比较困难的事情。在本文中我们将介绍一些常用的Bam文件打开和查看工具,并详细解析测序数据的格式和解读方法,帮助初学者快速掌握Bam文件的基础知识。
测序数据的格式解读方法
做了那么多测序数据,公司给了那么多文件,各个文件都是个啥?怎么有的文件连打开都打开不了,是我的编辑器有问题?(编辑器又是个什么玩意?)怎么有的文件打开的那么慢,咦,把我的编辑器都弄崩了。好吧,关闭重新打开,咦,怎么还是打不开?好吧,我放弃了。
了解了下面的介绍,可能你就能解决以上问题了。
下面主要介绍三种文件格式:1.fasta。2.fastq。3.sam。
1.Fasta/fa
我们的测序数据要往参考基因组比对吧,参考基因组呢,就是fasta格式,fasta格式应该比较熟悉,是这样子滴。

一般是一行染色体信息,接着下一行为序列信息。动物的参考基因组一般都比较大,但基本记事本都能打开。
2.fastq
这个是测序之后的原始数据,一般测序公司都会发给我们。可能是压缩文件,不过都一样了,我们打开一看,怎么看起来奇怪的,来,慢慢的学起来了。

这张图是不是很清楚了?什么,不清楚,好吧,我们再详细点。
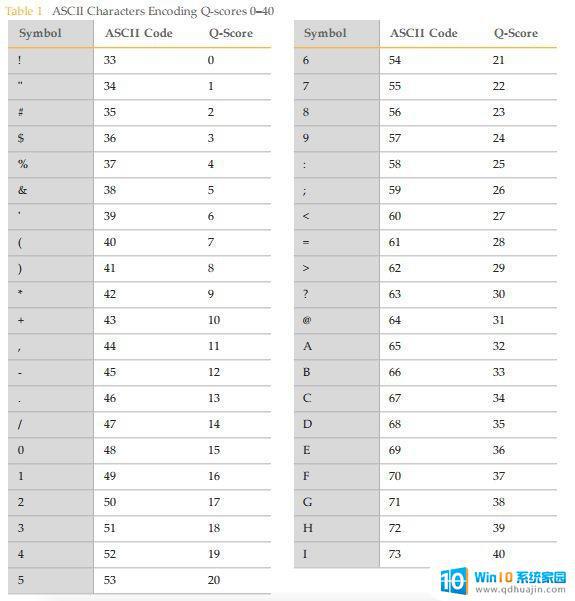
Fastq数据的行数是4的倍数,每四行表示一条read,这四行每行都代表着这条read的信息。第一行,以@起始,可以表示这条read的ID;第二行就是这条read的序列;第三行是描述行;第四行表示每个第二行中每个碱基对应的质量,怎么还有A、\等信息呢?第四行与碱基对应位置的字符代表了这个碱基的质量,其中A、\等都表示对应的碱基的质量。(字符是啥?A是个字符,\也是个字符)。其中上图中框出来的,C是碱基,E就是对应的碱基质量。那E是多少呢?再看这张图有和Q-score对应的得分,E就是36.
3.Sam/Bam
Sam/bam文件为比对后的文件,这个公司一般会给我们bam文件。大小相对较小,和sam的内容一样,只是被经过了压缩。再详细一点,Sam是标准比对文件格式。Sequence Alignment/Map format 的缩写,以tab分隔,可以用普通编辑器打开并查看。Bam文件是sam文件用bgzf方式压缩的二进制文件,不能用普通的编辑器打开,可以用samtools view查看。Sam文件和bam文件可以通过工具互相转换。接下来详细的了解sam格式。
Sam格式包括了3部分:1.文件头格式。2.比对格式。3.tag标记格式。
1.文件头格式:

其中每种type表示什么意思如下图
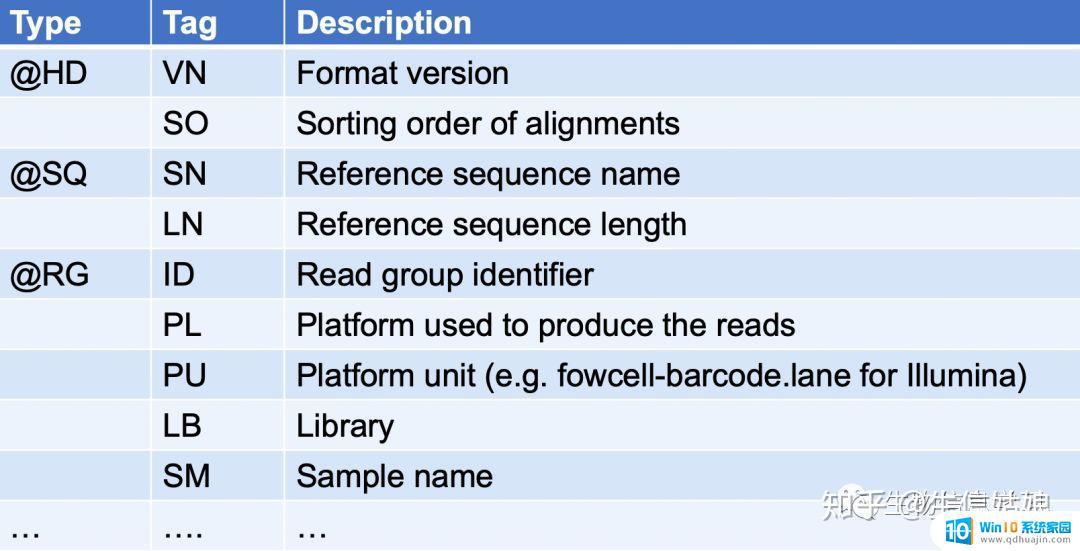
其中VN表示版本号;so表示排序方法,有按qname排序,有按位置排序;sn表示参考序列名字,ln参考序列长度;id表示readgroupid等。
2.比对格式
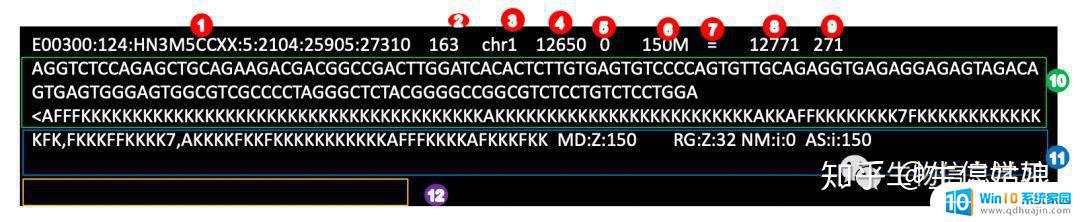
其中1-12分别和下图中的field对应

1.就是模板名字,可以认为是read的ID。2.flag这个下面会有详细讲解。3.参考序列的名字。4.mapping的参考基因组上的位置。5.mapping质量。6.cigar,这个下节可以详细讲解。7.R2对应的参考序列的名字。8.R2的位置。9.模板的长度。10.序列。11.每个base对应的质量。12.tag
除了这些测序常用的格式之外,还有vcf文件,bed文件,gtf,gff等,留着下次介绍吧。不早了,我要写bug去了(噢,不,debug)。
打开和查看bam文件需要使用专业的软件和工具进行操作,而对于测序数据格式的解读方法,需要具备一定的生物信息学基础知识和技能。只有深入学习和掌握相关的知识和技术,才能更好地理解和利用测序数据,并为生物学研究提供有价值的支持和帮助。
bam文件如何打开查看 测序数据的格式解读方法相关教程
热门推荐
电脑教程推荐
win10系统推荐
- 1 萝卜家园ghost win10 64位家庭版镜像下载v2023.04
- 2 技术员联盟ghost win10 32位旗舰安装版下载v2023.04
- 3 深度技术ghost win10 64位官方免激活版下载v2023.04
- 4 番茄花园ghost win10 32位稳定安全版本下载v2023.04
- 5 戴尔笔记本ghost win10 64位原版精简版下载v2023.04
- 6 深度极速ghost win10 64位永久激活正式版下载v2023.04
- 7 惠普笔记本ghost win10 64位稳定家庭版下载v2023.04
- 8 电脑公司ghost win10 32位稳定原版下载v2023.04
- 9 番茄花园ghost win10 64位官方正式版下载v2023.04
- 10 风林火山ghost win10 64位免费专业版下载v2023.04